Pathology and Laboratory Medicine
Severe bleeding due to an acquired FXIII inhibitor in an otherwise healthy patient
Spring 2024, Volume 22, Issue 2
Bianca Santonastaso ’24, Hannah Spector, Christine Cahill, Frank Akwaa, Jainulabdeen J. Ifthikharuddin, Gahyun Gim, Majed Refaai*
http://doi.org/10.47761/SYIQ6533
After being admitted to the hospital, the patient received one unit of packed red blood cells (pRBC). Further laboratory results the next morning showed significantly low FXIII levels (Table 1), and the patient continued to develop new and expanding ecchymosis and hematoma on his right back side over the next two days. Spontaneous bruising also appeared along his belt line and right groin, with mild tenderness on these areas (Figure 1). His FXIII levels continued to drop (from 37 to 4%) along with hemoglobin (Hgb) levels (from 7.5 to 6.7g/dL), but no other significant changes were observed. On the fourth and sixth day of the patient’s stay in the hospital, two more pRBC units and three doses of cryoprecipitate were administered to mitigate his symptoms. Moreover, the lack of bleeding history in this patient and the critically low FXIII levels raised concern for presence of an acquired inhibitor; however, an FXIII inhibitor failed to be detected despite laboratory diagnoses using two different FXIII functional assays and a modified mixing study. Bleeding greatly improved after administration of prednisone and FXIII concentrate drugs (Corifact, CSL Behring, King of Prussia, PA). On the sixth day, the patient was discharged, and prescribed aminocaproic acid and prednisone for presumed FXIII inhibitor.
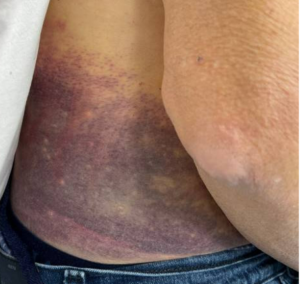
Figure 1: Right flank hematoma present upon first admission.
Three days later, he was readmitted with weakness, fatigue, dizziness, nausea, diffuse ecchymosis bruising, and swelling and numbness in his latissimus dorsi and lateral right thigh. He also reported right-sided leg pain and numbness in the distal right thigh. Lab tests revealed a FXIII level of 46% and a significantly low Hgb level of 8.1 g/dL. Further FXIII tests performed at a reference laboratory (ARUP) revealed <1% of normal activity, consistent with severe FXIII deficiency (Table 1).
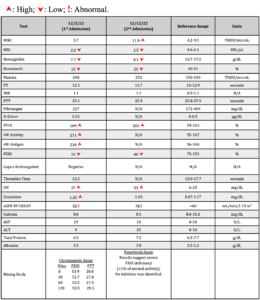
Table 1: Laboratory work-up for both admissions
An abdominal CT scan revealed hematoma along the abdominal wall musculature and the same swelling seen in the scapula upon his first admission. Throughout the following days in the hospital, the patient developed bilateral hemothorax (bleeding into the chest) and arterial bleeding in his thigh. To prevent further bleeding and injury, he underwent an angiogram with embolization procedure, receiving 2 pRBCs, 3 doses of cryoprecipitate, tranexamic acid (TXA), and multiple FXIII concentrate (40U/Kg) infusions (Figure 2).

Figure 2: FXIII levels (chromogenic) over both admissions
Over the next few days, intermittent FXIII concentrate infusions controlled the patient’s bleeding and maintained FXIII levels between 51-69%. Weekly Rituximab antibody medication, prednisone, and concentrated FXIII replacement therapy was prescribed, and the patient was discharged four days later once bleeding was maintained.
Four weeks after discharge, the patient’s aminocaproic acid treatment was discontinued. The first cycle of Rituximab was completed four months after discharge, while the prednisone was reduced from 40mg daily to 5mg biweekly. For the following six months, his FXIII levels maintained within normal limits; however, these changes in FXIII levels over the course of the treatment were directly correlated with immunotherapy and FXIII replacement administration prescribed (Figure 2). Although the patient tolerated a decrease in prednisone dosage to 5mg daily, he experienced a vast drop in FXIII levels once prednisone was reduced to biweekly doses. Once the steroid dosage was increased to 10mg daily and supplemented with additional FXIII concentrate, his FXIII levels immediately returned to baseline (Figure 2).
Conclusion:
The half-life of FXIII is approximately 11 to 14 days, which is useful in determining administration of specific drugs. It is supported in literature that FXIII levels above 5% are hemostatically adequate, but anything below 5%, especially at levels less than 1-3%, can result in severe bleedings. Although not identified serologically, the inhibitor was nonetheless present in this patient, indicated by significant bleeding, and clinical tests that showed persistently low FXIII levels, suboptimal response to cryoprecipitate and FXIII concentrate replacement therapy, and the favorable response to immunotherapy.
REVIEW
Background
The fibrin-stabilizing effect of Factor XIII (FXIII) was initially reported in 1923 by Barkin and Gasper, who demonstrated that fibrin clots were insoluble in weak bases [1]. In 1948, Laki and Lorand identified a “protein fibrin stabilizing factor” that was responsible for the formation of the fibrin clot [1]. A decade later, Lowey et al. purified and determined the mechanism of this factor [2]. In 1961, the first pediatric patient with impaired wound healing, abnormal scarring, severe bleeding, and decreased “protein fibrin stabilizing factor” was reported [1]. The role of this clot stabilizing factor was then recognized and designated by the International Committee on Blood Clotting Factors as “Factor XIII” [3].
In plasma, FXIII circulates as a heterotetramer composed of two catalytic A-subunits and two carrier B-subunits [4]. It is a multifunctional molecule known for its role in hemostasis, wound healing, angiogenesis, pregnancy maintenance, bone metabolism, and cardio protection [4,5]. FXIII deficiency can be either congenital or acquired, and may result in a number of complications such as delayed bleeding episodes, as it plays a critical role in stabilizing fibrin monomers in the final step of clot formation.
Congenital FXIII deficiency is a rare autosomal recessive disorder, estimated to affect one in two million people, with activity levels between 5-20% [6,7]. Heterozygous FXIII deficiency is usually asymptomatic, with an activity range of 50-70% [8]. In newborns, umbilical cord bleeding is a strong indication for diagnosis of congenital FXIII deficiency, and the most common presentation of the disorder. Other signs of FXIII deficiency in heterozygous cases include subcutaneous bleeding, intracranial hemorrhages, muscle hematomas, and severe bleeding [4,9].
Acquired FXIII deficiency is much less common than congenital deficiencies, and onset may be caused by increased consumption, either from bleeding, antibodies, disseminated intravascular coagulation (DIC), or decreased synthesis from liver failure or antibodies. Immune-related disorders such as leukemia, liver disease, rheumatoid arthritis, and systemic lupus erythematosus are correlated with decreased FXIII activity [4,10]. Furthermore, the acquired deficiency could be attributed to long-term treatment and utilization of drugs such as isoniazid penicillin, phenytoin, practolol, and amiodarone [4,11]. The manifestation of these anti-FXIII antibodies may neutralize activated FXIII, increase FXIII clearance, or interfere with FXIII-fibrin interaction [7].
Diagnosis
FXIII deficiency is characterized by prolonged bleeding episodes and impaired wound healing in soft tissues [12]. When diagnosing a bleeding disorder such as FXIII deficiency, it is important to collect the patient’s personal and family history. This will provide a means to ascertain whether the disorder is acquired or congenital in nature. Acquired deficiency may be suspected if there is no relevant bleeding history [4].
Patients with congenital FXIII deficiencies typically present with umbilical cord bleeding [4]. Due to the role FXIII plays in pregnancy, recurrent spontaneous miscarriage and postpartum hemorrhage is relatively common [4,6]. Although incredibly rare, central nervous system bleeding is the leading cause of death in these patients [6]. Severe bleeding may also occur in patients who become resistant to FXIII replacement therapy [12]. As a whole, most patients with congenital deficiency range between 5-20% FXIII activity and have a variety of bleeding symptoms [7].
Up to 70% of patients with acquired FXIII deficiencies present with soft tissue hematomas [4]. With newly developed antibodies, many patients experience a drastic plummet in FXIII activity and consequently, life-threatening intracranial, intrathoracic, or intraperitoneal hemorrhages [4,12]. These bleeding episodes may also present as mucocutaneous and intramuscular bleeding [12]. Nevertheless some reports note that symptoms of acquired FXIII deficiency have no correlation with residual FXIII activities [7].
Due to the rarity of both congenital and acquired FXIII deficiency, other bleeding disorders, such as von Willebrand disease (vWD), must be ruled out to ensure proper diagnosis. In one study, disseminated intravascular coagulation was most commonly confused with FXIII deficiency; however, DIC normally presents as a decrease in more than one coagulation factor from increased consumption [11].
Laboratory Evaluation
Most routine coagulation testing, including prothrombin time (PT), activated partial thromboplastin time (aPTT), thrombin time (TT), and platelet count show normal levels, making FXIII deficiency difficult to diagnose [13,14]. Moreover, since FXIII is responsible for stabilizing the clot formed throughout the coagulation cascade, neither the PT or aPTT monitor the clot beyond initial formation and both appear within normal limits even in FXIII deficiency cases. In the past, clot solubility tests were utilized to screen for FXIII deficiency, but have since been discontinued due to the high rates of false negatives in milder forms of the deficiency [4,7].
Since FXIII deficiency is difficult to detect, other testing options include a variety of assays: photometric, incorporation, fluorometric, Bethesda, clot-based inhibitor assays, as well as antigen immunoassays and mixing studies [4]. Some studies propose that thrombin generation and thromboelastometry studies could be beneficial in monitoring hemostasis and clot strength [12]. Other studies suggest utilizing FXIII antigen assays to quantify the amount of FXIII in circulation based on subunit A and B, as well as the AB complex; however, there is no consensus on what level of FXIII indicates FXIII deficiency [4,14].
Treatment/Management
Treatments for FXIII disorders are difficult to standardize given the rarity of FXIII deficiency [4], but many patients with acquired deficiencies are treated with recombinant FXIII concentrates and immunosuppressants. However, congenital FXIII deficiency may also develop inhibitors in response to FXIII concentrate doses.
Once FXIII deficiency has been established, it is crucial to begin preventative prophylactic treatment due to the variable nature of the associated symptoms [4]. Though clinical guidelines are limited, many utilize a combination of fresh frozen plasma (FFP), cryoprecipitate, FXIII concentrates, and/or antifibrinolytic agents to restore FXIII levels [10,14]. Some argue FXIII concentrate should not be used in perioperative care due to insufficient evidence of benefits, and recommend only using it for life-threatening cases of bleeding [7,14]. Alternatively, antifibrinolytics, tranexamic acid, and aminocaproic acid treatments are relatively commonly used when treating FXIII deficiency [11]. Tranexamic acid (TXA) is capable of facilitating antifibrinolytic mechanisms, mimicking FXIII, which is capable of both antifibrinolytic properties and fibrin stabilization [11]. In cases of immune-acquired inhibitors, immunosuppressants are a necessity in preventing recurrent bleeding events and eliminating antibodies [10,12]. This may include steroids, cyclophosphamide, or Rituximab, which have all been shown to eradicate FXIII inhibitors [4,10]. In particular, Rituximab is an anti-CD20 monoclonal antibody used in approximately 24% of acquired FXIII deficiency cases since 2005 [11]. An alternative option is plasma exchange therapy, which removes inhibitors from circulation [11]. However, even with therapy, both patients with congenital or acquired deficiencies can experience refractory bleeding [7].
With no consensus on a standard FXIII level, there are various recommendations ranging from levels above 0.5%, 2%, 5%, 10%, to even as high as 30% to maintain hemostasis [4,14,15]. Hemostatic levels can also be achieved with FXIII concentrate infusions, which are recommended at the same dosage as FFP, every 4-6 weeks [4,16]. It is recommended to maintain elevated hemostatic levels in severe acute bleeding cases, and for major surgeries at levels of 50% or higher [4]. Historically, greater than 5% of FXIII was adequate for preventative prophylaxis, though newer case reports are finding that these levels are not always satisfactory to prevent spontaneous bleeding, as levels can present in a number of ways.
References
- Mangla, A., Hamad, H., & Kumar, A. (2022). Factor XIII Deficiency. In StatPearls. StatPearls Publishing.
- Chandrasekhar, N., Osbahr, A., & Laki, K. (1964). THE MODE OF ACTION OF THE LAKI-LORAND FACTOR IN THE CLOTTING OF FIBRINOGEN. Biochemical and biophysical research communications, 15(2), 182–187. https://doi.org/10.1016/0006-291x(64)90321-3
- Muszbek, L., Ariëns, R. A., Ichinose, A., & ISTH SSC SUBCOMMITTEE ON FACTOR XIII (2007). Factor XIII: recommended terms and abbreviations. Journal of thrombosis and haemostasis: JTH, 5(1), 181–183. https://doi.org/10.1111/j.1538-7836.2006.02182.x
- Dorgalaleh, A., Kazemi, A., Zaker, F., Shamsizadeh, M., Rashidpanah, J., & Mollaei, M. (2016). Laboratory Diagnosis of Factor XIII Deficiency, Routine Coagulation Tests with Quantitative and Qualitative Methods. Clinical laboratory, 62(4), 491–498. https://doi.org/10.7754/clin.lab.2015.150619
- Muszbek, L., Yee, V. C., & Hevessy, Z. (1999). Blood coagulation factor XIII: structure and function. Thrombosis research, 94(5), 271–305. https://doi.org/10.1016/s0049-3848(99)00023-7
- Motlagh, H., Dorgalaleh, A., Tabibian, S., Naderi, M., & Zaker, F. (2022). Noninvasive prenatal diagnosis of congenital factor XIII deficiency in Iran. Blood coagulation & fibrinolysis: an international journal in haemostasis and thrombosis, 33(3), 167–170. https://doi.org/10.1097/MBC.0000000000001121
- Beckman, J. D., Kasthuri, R. S., Wolberg, A. S., & Ma, A. D. (2018). Challenges in diagnosis and management of acquired factor XIII (FXIII) inhibitors. Haemophilia : the official journal of the World Federation of Hemophilia, 24(6), e417–e420. https://doi.org/10.1111/hae.13603
- Kohler, H. P., Ichinose, A., Seitz, R., Ariens, R. A., Muszbek, L., & Factor XIII and Fibrinogen SSC Subcommittee Of The ISTH (2011). Diagnosis and classification of factor XIII deficiencies. Journal of thrombosis and haemostasis: JTH, 9(7), 1404–1406. https://doi.org/10.1111/j.1538-7836.2011.04315.x
- Naderi, M., Dorgalaleh, A., Alizadeh, S., Tabibian, S., Hosseini, S., Shamsizadeh, M., & Bamedi, T. (2014). Clinical manifestations and management of life-threatening bleeding in the largest group of patients with severe factor XIII deficiency. International journal of hematology, 100(5), 443–449. https://doi.org/10.1007/s12185-014-1664-1
- Chou, S. C., Lin, C. Y., Yen, C. T., Hsieh, H. N., Huang, Y. C., Li, K. J., Lin, S. W., & Shen, M. C. (2021). Acquired FXIII inhibitor: Patient characteristics and treatment outcome, a case series in Taiwan. Journal of the Formosan Medical Association = Taiwan yi zhi, 120(1 Pt 2), 411–414. https://doi.org/10.1016/j.jfma.2020.05.032
- Ichinose, A., & Japanese Collaborative Research Group on AH13 (2017). Autoimmune acquired factor XIII deficiency due to anti-factor XIII/13 antibodies: A summary of 93 patients. Blood reviews, 31(1), 37–45. https://doi.org/10.1016/j.blre.2016.08.002
- Marco, A., & Marco, P. (2021). Autoimmune Acquired Factor XIII Deficiency: A Case Report. Journal of blood medicine, 12, 63–68. https://doi.org/10.2147/JBM.S288634
- Dorgalaleh, A., & Rashidpanah, J. (2016). Blood coagulation factor XIII and factor XIII deficiency. Blood reviews, 30(6), 461–475. https://doi.org/10.1016/j.blre.2016.06.002
- Kleber, C., Sablotzki, A., Casu, S., Olivieri, M., Thoms, K. M., Horter, J., Schmitt, F. C. F., Birschmann, I., Fries, D., Maegele, M., Schöchl, H., & Wilhelmi, M. (2022). The impact of acquired coagulation factor XIII deficiency in traumatic bleeding and wound healing. Critical care (London, England), 26(1), 69. https://doi.org/10.1186/s13054-022-03940-2
- Duque, P., Chasco-Ganuza, M., Ortuzar, A., Almaraz, C., Terradillos, E., Perez-Rus, G., & Pascual, C. (2022). Acquired FXIII Deficiency is Associated with High Morbidity. Thrombosis and haemostasis, 122(1), 48–56. https://doi.org/10.1055/a-1481-2733
- Cojutti, P. G., Zanon, E., Pasca, S., Pea, F., & Italian FXIII Study Group (2022). Real-Life Population Pharmacokinetics of Recombinant Factor XIII and Dosing Considerations for Preventing the Risk of Bleeding in Patients with FXIII Congenital Deficiency. Clinical pharmacokinetics, 61(4), 505–513. https://doi.org/10.1007/s40262-021-01079-x
About the Author
Bianca Santonastaso is a pre-medical student graduating in May of 2024 with a Bachelor of Science degree in Biochemistry and a minor in Clinical Psychology. Her research with the Hemostasis and Thrombosis Research Laboratory in the Department of Pathology at the University of Rochester Medical Center (URMC) began in 2022. As a laboratory technician, she has both led and contributed to projects involving the assessment of patented Platelet Modified Lysate (PML) blood product applications on animals, Engraftment Syndrome/Pre-Engraftment Syndrome transfusion needs, URMC blood product utilization, and special coagulation laboratory instrument evaluations for FDA-clearance, among additional research studies and clinical involvement.
Cite this Article
Santonastaso, B., Spector, H., Cahill, C., Akwaa, F., Ifthikharuddin, J. J., Gim, G., Refaai, M. (2024). Severe bleeding due to an acquired FXIII inhibitor in an otherwise healthy patient. University of Rochester, Journal of Undergraduate Research, 22(2). http://doi.org/10.47761/SYIQ6533
JUR | Creative Commons Attribution BY 4.0 International License![]()