THESIS in Microbiology & Immunology
Estrogen Receptor α (ERα) Signaling and Tumor Cell-Derived Factors Mediate Pro-Tumor Neutrophil Activation in the Setting of Lymphangioleiomyomatosis (LAM)
Fall 2024, Volume 23, Issue 1
Katherine O’Leary ’25 (T5), Erin Gibbons, Briaunna Minor, Thomas Henson, Stephen R Hammes*
https://doi.org/10.47761/WZIH8024
Introduction
Lymphangioleiomyomatosis (LAM) is a rare multisystem disease that predominantly manifests in the lungs. [1]. It is characterized by the abnormal growth of smooth muscle tumors, which leads to cyst formation in the lungs and the progressive loss of respiratory function [1]. Common symptoms include shortness of breath, coughing, and chest pain, with impacted individuals also being susceptible to recurrent lung collapse. Diagnosis is typically confirmed through a biopsy of lung tissue or lymph nodes, as pulmonary function tests may appear normal in earlier stages of the disease [2, 3]. Computed tomography (CT) scans are also helpful when diagnosing LAM, as they visualize cystic changes in the lungs [2]. Extrapulmonary manifestations of LAM include the presence of benign smooth muscle tumors in the kidney or uterus, lymphadenopathy, and large cystic lymphatic masses (lymphangioleiomyomas) [2].
There are a few notable features of LAM that inform ongoing investigations into its mechanisms. First, genetically, LAM is caused by mutations in the tuberous sclerosis complex (TSC) genes, TSC1 and TSC2, with most cases linked to inactivating mutations in TSC2 [4]. These mutations may occur sporadically or as inherited germline mutations in tuberous sclerosis. Regardless of origin, defective TSC signaling leads to hyperactivation of the mammalian target of rapamycin complex 1 (mTORC1) promoting excessive cell growth and proliferation. The main treatment for this disease includes rapamycin-like drugs which target the hyperactivity of mTORC1 to attenuate disease progression and provide symptom relief [2]. In severe cases of respiratory decline secondary to disease progression, lung transplantation may be required. Some patients experience adverse effects from rapalogs or recurrence of LAM following lung transplantation, highlighting the need for further investigations into the disease mechanism to identify potential therapeutic targets [2].
Second, LAM is considered to be a metastatic disease, as LAM cells have been found in the blood and lymph vasculature of patients. Recurrence in transplanted lungs is also common [1]. Third, LAM exhibits a marked female sexual dimorphism in disease presentation, as it predominantly occurs in biological females and rarely affects males (less than 10 cases of significant disease reported). When it does present in males, it generally has no to mild symptoms [3]. Finally, LAM is an estrogen-sensitive disease, causing it to worsen during reproductive years, with pregnancy, and with oral contraception use, though improving after menopause [1].
The estrogen responsiveness of LAM has been the subject of studies aiming to elucidate the potential benefits of hormone-targeting therapies to combat this disease. For instance, prior preclinical studies have shown that estrogen enhances lung colonization of TSC2-null tumor cells, derived from Eker rat leiomyomas (ELT3), in a SCID.NOD metastasis model [3, 5]. Using the (Progesterone Receptor) PRCre/+ TSC2fl/fl uterine-specific TSC2-null mouse model, which develops LAM-like tumors in the myometrium, our previous work has demonstrated that estrogen removal causes tumor regression [6]. These in vivo data, along with others, highlights that LAM-like tumors are not just estrogen-responsive, but estrogen-dependent [5-8]. Interestingly, while in vivo response to estradiol is significant, recent studies show mild responsiveness of TSC2-null tumor cells to estradiol stimulation in vitro. Using a novel in vitro model of TSC2-null cells derived from the uterine tumors of the TSC2fl/fl PRCre/+ mouse (LTM3 cells), we previously found that these LAM-like tumor cells were transcriptionally reactive to estradiol stimulation, but did not augment pro-tumorigenic functions such as migration, invasion, or proliferation in response to estradiol treatment [3]. With these results in mind, recent studies have shifted to investigating the estrogen sensitivity of other cellular components of the tumor microenvironment to elucidate the mechanisms of estrogen-mediated LAM progression.
LAM tumors have a diverse microenvironment consisting of fibroblasts, adaptive immune cells (B and T lymphocytes), and innate immune cells (monocytes, macrophages, dendritic cells, mast cells, and neutrophils) [1, 9]. Similar to other malignancies, crosstalk within the LAM microenvironment enables these non-tumor cells to support tumor growth through immunosuppressive and direct pro-tumorigenic mechanisms [1, 3]. Interestingly, recent single-cell RNA characterization of LAM-diseased lungs revealed that CD45-expressing immune cells expressed a high level of ESR1 (the gene encoding ERα) much like the LAM tumor cells [10]. These data substantiated previously reported modulations in immune cell-related mRNAs in the preclinical TSC2fl/fl PRCre/+ murine model [3, 6]. Specifically, estradiol stimulation in uterine tissues led to increased expression of neutrophil elastase (NE), a serine protease reported to be associated with tumor progression for other malignancies [6, 11, 12]. Further immunophenotyping of myometrial tumors in TSC2fl/flPRCre/+ mice revealed significant increases in CD11b+Ly6CloLy6G+ (neutrophil) cell infiltration into uteri burdened with LAM-like tumors compared to tumor-naïve uteri [11]. These neutrophils were also identified as potent regulators of TSC2-null tumor growth in the myometrium, as evidenced by marked tumor growth reduction following the depletion of myeloid cells, inhibition of their recruitment, and inhibition of neutrophil-derived neutrophil elastase (NE) [11].
It is well-established that neutrophils play a crucial role in fighting infections [13, 14]. However, research suggests that environmental stimuli can modulate neutrophil functions, allowing them to utilize a diverse repertoire of functions that, in some instances, can support disease progression [15]. In cancer, neutrophils have been shown to promote tumor growth by releasing a variety of different factors that stimulate tumor cells directly, such as Mmp9 and ELANE, or by supporting evasion of cytotoxic immune cells via immunosuppressive means through Nos2, Cd274, and S100a9 [12, 15-18]. Given the evidence implicating both estrogen and neutrophils as drivers of LAM-like tumor progression, understanding how estrogens influence neutrophil accumulation and function in the LAM tumor microenvironment is critical. Our lab has previously demonstrated that estradiol increases neutrophil levels, facilitating greater colonization of LAM-like tumors in the lungs of SCID.NOD mice [3]. Neutrophil plasticity, which allows for the versatility of neutrophil functions and phenotypes,likely supports neutrophil and tumor cell clusters [19]. Since neutrophils play a role in estrogen-mediated lung colonization, the influence of estrogen on neutrophil plasticity is an important feature of neutrophils that must be explored when looking at the LAM microenvironment.
Based on all the data mentioned, we hypothesize that estradiol signaling through estrogen receptor-alpha on neutrophils is promoting a pro-tumorigenic immunosuppressive phenotype, which in turn supports the progression of LAM. Supporting this, our results indicate that ERα positively influences the expression of neutrophil elastase (Elane) and nitric oxide synthase 2 (Nos2), markers of neutrophil activation, to support tumor progression.

Figure 1. The model of crosstalk between Neutrophils and LAM cells is modulated by estrogen.
We hypothesize that estrogen supports the crosstalk between LAM tumor cells and neutrophils in the tumor microenvironment, which in turn supports the progression of LAM. LAM tumor cells secrete tumor-derived factors that alter the neutrophil phenotype, making the neutrophils express pro-tumorigenic and immunosuppressive markers that support the tumor cells.
Materials and Methods
Mouse Studies.
Mouse studies were performed following AALAC guidelines and approved by the University of Rochester Committee on Animal Resources (UCAR).
Tumor Cell Culture and Reagents.
The LAM-like TSC2-null Myometrial (LTM3) cells are derived from TSC2-null myometrial tumors of TSC2fl/fl PRCre/+ mice [4]. LTM3 cells were maintained in DF8 media with 10% FBS and were incubated in incubators at 37ºC in air humidified with 5% CO2 and passaged every 4-5 days. Tumor-conditioned media (TCM) was collected after culturing the LTM3 cells to confluency in regular DF8 with 10% FBS.
Neutrophil Isolation.
Bone marrow was harvested from both the tibia and femur of three 18-week-old C57BL/6J (#000664, Jackson Laboratory) or B6N(Cg)-ESR1tm4.2Ksk/J ERα-null (#026176, Jackson Laboratory) mice and pooled for neutrophil isolation. The bone marrow was then placed into ACK lysis buffer to remove blood cells, neutralized, passed through a 40mm filter, and resuspended in FACs buffer. Single-cell suspensions of filtered bone marrow were stained with anti-Ly6G microbeads (#130120337, Miltenyi), and neutrophils were isolated according to the protocol for MS column MACS positive-selection separation (#130042201, Miltenyi). Once separated, neutrophils were counted and resuspended in the appropriate media at 1×106 neutrophils per mL.
Neutrophil Stimulation Cultures.
1×106 neutrophils per mL were seeded into 6-well plates with either DF8 media supplemented with 10% FBS or TCM. Stock Fulvestrant (#ICI182780, MedChemExpress), an estrogen receptor antagonist, was diluted to a working concentration of 1 mM using DMSO and stored in 10 mL aliquots at -80°C. Neutrophils were stimulated with 100 nM Fulvestrant or vehicle (DMSO). Cultures were incubated for 24 hours at 37°C in air humidified with 5% CO2.
Western Blot.
In preparation for Western Blot, samples were homogenized in RIPA buffer (#89900, Thermo Fisher Scientific, Waltham, MA) supplemented with 1×Halt™ protease and phosphatase-inhibitor cocktail (7843036, Thermo Fisher Scientific) and mixed with 4x-sample buffer. Samples were mixed with 4x sample buffer, boiled for five minutes, and were then loaded into gradient polyacrylamide gels to separate protein (4561084, Bio-Rad, Hercules, CA). Samples were then transferred to PVDF membranes (#1620177, Bio-Rad), and blots were blocked using 5% milk in TBST. The primary antibody was 1:1000 Erα (8644S, Cell Signaling). Secondary antibodies included 1:4000 Goat anti-rabbit (#1706515, Bio-Rad). Clarity Western ECL Substrate (1705062, Bio-Rad) was used to detect the blot.
Real-Time Quantitative PCR.
Total RNA was extracted using the RNeasy Plus Mini Kit (74134, QIAGEN, Hilden, Germany). RT-qPCR reactions used qScript RT-qPCR Tough Mix (89236-672, Quantabio, Beverly, MA) and TaqMan primers (Applied Biosystems, Waltham, MA). The StepOne plus Real-Time PCR system (Applied Biosystems) was used. The Taqman Primers used were mouse GAPDH (loading control, Mm99999915_g1), mouse ELANE (Mm00469310_m1), mouse CXCR2 (Mm00438258_m1), mouse Nos2 (Mm00440502_m1), mouse IRF8 (Mm00492567_m1), mouse Cd274 (Mm03048248_m1), mouse S100a9 (Mm00656925_m1), and mouse Tgfb1 (Mm01178820_m1). Samples were tested in duplicates for the target gene expression. After, mRNA threshold cycle (ΔΔCT) values were normalized to GAPDH levels and represented the relative mRNA.
Statistical Analysis.
GraphPad PRISM 10 software was then used to express these results. Each probe was normalized to GAPDH levels for the specific day it was run, and then relative values were graphed. Unpaired t-tests were performed to determine the significance between the mean CT values, and differences in the means were compared using Tukey’s multiple comparisons test. Significance was denoted by ns (p > 0.05), * (p < 0.05), ** (p < 0.01), *** (p < 0.001).
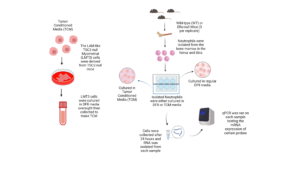 Figure 2. Schematic of Experiment. Tumor-conditioned media was derived from LAM-like TSC2-null Myometrial cells (LTM3) from TSC2-null mice. LTM3 cells were cultured overnight, then the media was collected to create the tumor-conditioned media condition. Neutrophils were harvested from wild-type (WT) and ERα-null (KO) mice and cultured in DF8 or TCM. In later experiments, the neutrophils were either cultured in DF8 with Vehicle (Veh) or Fulvestrant (Ful).
Figure 2. Schematic of Experiment. Tumor-conditioned media was derived from LAM-like TSC2-null Myometrial cells (LTM3) from TSC2-null mice. LTM3 cells were cultured overnight, then the media was collected to create the tumor-conditioned media condition. Neutrophils were harvested from wild-type (WT) and ERα-null (KO) mice and cultured in DF8 or TCM. In later experiments, the neutrophils were either cultured in DF8 with Vehicle (Veh) or Fulvestrant (Ful).
Results
Neutrophil Elastase (Elane) and Nitric Oxide Synthase 2 (Nos2) expression in neutrophils from the bone marrow are enhanced by ERα signaling.
Prior work has shown that estradiol stimulation in uterine tissues increased the expression of Neutrophil Elastase (Elane), which has pro-tumorigenic properties [6, 11, 12]. Additionally, Nitric Oxide Synthase 2 (Nos2) expression in cancer cells predicts poor outcomes, as it increases chemoresistance, metastasis and has immunosuppressive functions [17]. Both markers are predicted to be expressed by neutrophils in the presence of estrogen signaling in LAM. In this study, we used real-time quantitative polymerase chain reaction (RT-qPCR) to measure the mRNA expression of the genes Elane and Nos2 in both wild-type and ERα knockout mice in two conditions: DF8 media and tumor-conditioned media (TCM). Our results indicate that the expression of both Elane (Figure 3a) and Nos2 (Figure 3b) was dependent on ERα signaling in neutrophils. For Elane expression (Figure 3a), significant difference was observed between the WT and KO mice in DF8 conditions (*p < 0.05), while the difference in TCM was trending towards significance (p = 0.0807). For Nos2 expression (Figure 3b), there were significant differences between the baseline expression in WT and KO mice in both the DF8 and TCM conditions (*p < 0.05). These baseline differences in Elane and Nos2 indicate that ERα signaling increases the expression of these genes, potentially promoting a pro-tumorigenic neutrophil phenotype.
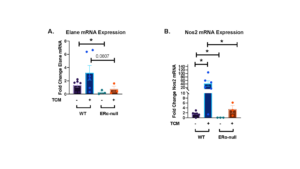 Figure 3. Neutrophil Elastase (Elane) and Nitric Oxide Synthase 2 (Nos2) mRNA expression are influenced by estrogen signaling in both DF8 media and tumor-conditioned media (TCM). Neutrophil Elastase (Elane) mRNA expression (a) and Nitric Oxide Synthase 2 (Nos2) mRNA expression (b) were measured in cultured neutrophils taken from the bone marrow of either either wild-type (C57BL/6J) or Erα null mice. Neutrophils were cultured in either DF8 media or tumor-conditioned media (TCM). For wild-type mice, n = 6, and for Erα null mice, n = 3. Each data point represents the mean of three biological replicates, with two technical replicates run for each. Data are represented as means ± SEM. Unpaired t-tests determined the significance between conditions. ns p > 0.05, *p < 0.05.
Figure 3. Neutrophil Elastase (Elane) and Nitric Oxide Synthase 2 (Nos2) mRNA expression are influenced by estrogen signaling in both DF8 media and tumor-conditioned media (TCM). Neutrophil Elastase (Elane) mRNA expression (a) and Nitric Oxide Synthase 2 (Nos2) mRNA expression (b) were measured in cultured neutrophils taken from the bone marrow of either either wild-type (C57BL/6J) or Erα null mice. Neutrophils were cultured in either DF8 media or tumor-conditioned media (TCM). For wild-type mice, n = 6, and for Erα null mice, n = 3. Each data point represents the mean of three biological replicates, with two technical replicates run for each. Data are represented as means ± SEM. Unpaired t-tests determined the significance between conditions. ns p > 0.05, *p < 0.05.
Programmed Cell Death Ligand 1 (PD-L1, Cd274) expression is mediated by the presence of tumor-conditioned media, rather than ERα signaling.
Programmed Cell Death Ligand 1 (PD-L1), also known as Cd274, is a checkpoint inhibitor that binds to PD-1 receptors on T cells, preventing T cells from carrying out anti-tumor functions [18, 20]. Prior studies have shown that Cd274 is expressed by immune cells, including neutrophils in tumor microenvironments, and inhibits anti-tumor T cell functions to promote tumor progression [18]. In this study, our results demonstrate that Cd274 mRNA expression (Figure 2a) increased in the presence of TCM, regardless of whether the cells were isolated from WT or ERα-null mice. The difference in expression between DF8 and TCM conditions for the WT mice was trending towards significance (p = 0.0787).In ERa-null mice, Cd274 expression was significantly elevated in the presence of TCM (*p < 0.05). There was also a slight but significant increase in the expression of Cd274 mRNA in the KO mice in DF8 media, indicating that estrogen signaling may decrease the expression of Cd274. However, there was no significant difference in the TCM condition between both mice. (p > 0.05).
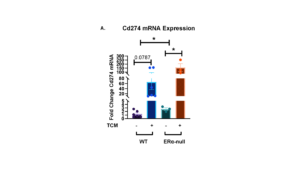 Figure 4. Increases in Programmed Cell Death Ligand 1 (PD-L1), also known as Cd274 mRNA expression is tumor-conditioned media mediated in both wild-type and ERα-null (KO) mice. PD-L1 mRNA expression (a), was measured for all cultured neutrophils taken from the bone marrow from either wild-type (C57BL/6J) (n = 6) or Erα null mice (n = 3). Neutrophils were cultured in either DF8 media or tumor-conditioned media (TCM). Each data point represents the mean of three biological replicates, with two technical replicates performed for each.The data plotted is represented as mean ± SEM. Unpaired t-test determined the significance between conditions with and without the presence of estrogen receptor α. ns p > 0.05, *p < 0.05, ** p < 0.01, *** p < 0.001.
Figure 4. Increases in Programmed Cell Death Ligand 1 (PD-L1), also known as Cd274 mRNA expression is tumor-conditioned media mediated in both wild-type and ERα-null (KO) mice. PD-L1 mRNA expression (a), was measured for all cultured neutrophils taken from the bone marrow from either wild-type (C57BL/6J) (n = 6) or Erα null mice (n = 3). Neutrophils were cultured in either DF8 media or tumor-conditioned media (TCM). Each data point represents the mean of three biological replicates, with two technical replicates performed for each.The data plotted is represented as mean ± SEM. Unpaired t-test determined the significance between conditions with and without the presence of estrogen receptor α. ns p > 0.05, *p < 0.05, ** p < 0.01, *** p < 0.001.
C-X-C Motif Chemokine Receptor 2 (Cxcr2) mRNA expression is decreased in the presence of ERα signaling.
Previous studies have shown that C-X-C Motif Chemokine Receptor 2 (Cxcr2) is upregulated in various cancers and is associated with poorer prognosis, indicating some pro-tumorigenic effects such as promoting tumor cell proliferation [21]. Cxcr2 affects neutrophil maturation and function and is expressed by neutrophils in both mice and humans [22]. In addition, previous work from our lab has shown that Cxcr2 inhibition decreases uterine weight in the uterine-specific TSC2-null mouse model [11]. In this study, our results indicated that Cxcr2 expression is greatly decreased in the presence of ERα signaling in both DF8 and TCM conditions. There was a significant decrease in expression between the neutrophils cultured in DF8 media (***p < 0.001), as well as between the neutrophils cultured in TCM (*p < 0.05) in the wild-type mice. There was no significant difference in Cxcr2 expression between the DF8 and TCM conditions in either mouse model, suggesting that mRNA expression is not affected by TCM but is inhibited by ERα signaling possibly.
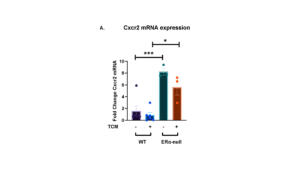 Figure 5. C-X-C Motif Chemokine Receptor 2 (Cxcr2) mRNA expression is not decreased in the presence of ERα signaling in both wild-type and knockout mice. C-X-C Motif Chemokine Receptor 2 (Cxcr2) mRNA expression (a) was measured for all cultured neutrophils taken from the bone marrow from either wild-type (C57BL/6J) or Erα null mice. Neutrophils were cultured in either DF8 media or tumor-conditioned media (TCM). For wild-type mice, n = 6, while for Erα null mice, n = 3. For each point, three biological replicates were used and two technical replicates were run. The data plotted is represented as means ± SEM. Unpaired t-test determined the significance between conditions with and without the presence of estrogen receptor α. ns p > 0.05, *p < 0.05, ** p < 0.01, *** p < 0.001
Figure 5. C-X-C Motif Chemokine Receptor 2 (Cxcr2) mRNA expression is not decreased in the presence of ERα signaling in both wild-type and knockout mice. C-X-C Motif Chemokine Receptor 2 (Cxcr2) mRNA expression (a) was measured for all cultured neutrophils taken from the bone marrow from either wild-type (C57BL/6J) or Erα null mice. Neutrophils were cultured in either DF8 media or tumor-conditioned media (TCM). For wild-type mice, n = 6, while for Erα null mice, n = 3. For each point, three biological replicates were used and two technical replicates were run. The data plotted is represented as means ± SEM. Unpaired t-test determined the significance between conditions with and without the presence of estrogen receptor α. ns p > 0.05, *p < 0.05, ** p < 0.01, *** p < 0.001
Transforming Growth Factor Beta 1 (Tgfb1), S100 Calcium Binding Protein A9 (S100a9), and Interferon Regulatory Factor 8 (Irf8) mRNA expression are not affected by the presence of ERα or tumor-conditioned media (TCM).
All of these markers: Tgfb1, S100a9, and Irf8, are known to be pro-tumorigenic or immunosuppressive markers. Tgfb1 is highly expressed in the setting of some cancers and supports the growth and metastasis of tumor cells [23]. S100a9 is involved in the inflammatory pathways and migration in multiple types of myeloid cells [24]. Finally, Irf8 is a transcription factor that contributes to the development of myeloid cells and is involved in the inflammatory response [25]. Our results indicated that ERα signaling does not affect the mRNA expression of any of these genes. There were no significant increases or decreases between the mouse models or culture conditions for Tgfb1 (Figure 6a), S100a9 (Figure 6b), or Irf8 (Figure 6c).
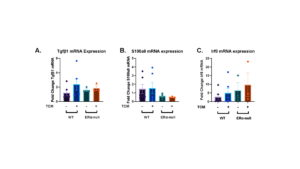 Figure 6. Transforming Growth Factor Beta 1 (Tgfβ1), S100 Calcium Binding Protein A9 (S100a9), and Interferon Regulatory Factor 8 (Irf8) mRNA expression does not change in both WT and KO mouse models cultured in DF8 or TCM. Transforming Growth Factor Beta 1 (Tgfβ1) (a), S100 Calcium Binding Protein A9 (S100a9) (b), and Interferon Regulatory Factor 8 (Irf8) (c) mRNA expression was measured for all cultured neutrophils taken from the bone marrow from either wild-type (C57BL/6J) or Erα null mice. Neutrophils were cultured in either DF8 media or tumor-conditioned media (TCM). For wild-type mice, n = 6, while for Erα null mice, n = 3. For each point, three biological replicates were used and two technical replicates were run. The data plotted is represented as means ± SEM. Unpaired t-test determined the significance between conditions with and without the presence of estrogen receptor α. ns p > 0.05, *p < 0.05, ** p < 0.01, *** p < 0.001.
Figure 6. Transforming Growth Factor Beta 1 (Tgfβ1), S100 Calcium Binding Protein A9 (S100a9), and Interferon Regulatory Factor 8 (Irf8) mRNA expression does not change in both WT and KO mouse models cultured in DF8 or TCM. Transforming Growth Factor Beta 1 (Tgfβ1) (a), S100 Calcium Binding Protein A9 (S100a9) (b), and Interferon Regulatory Factor 8 (Irf8) (c) mRNA expression was measured for all cultured neutrophils taken from the bone marrow from either wild-type (C57BL/6J) or Erα null mice. Neutrophils were cultured in either DF8 media or tumor-conditioned media (TCM). For wild-type mice, n = 6, while for Erα null mice, n = 3. For each point, three biological replicates were used and two technical replicates were run. The data plotted is represented as means ± SEM. Unpaired t-test determined the significance between conditions with and without the presence of estrogen receptor α. ns p > 0.05, *p < 0.05, ** p < 0.01, *** p < 0.001.
Treating neutrophils with Fulvestrant does not significantly alter mRNA expression in Neutrophil Elastase (Elane), in Programmed Cell Death Ligand 1 (PD-L1), also known as Cd274, Transforming Growth Factor Beta 1 (Tgfβ1), S100 Calcium Binding Protein A9 (S100a9).
All of the markers mentioned above possess either pro-tumorigenic or immunosuppressive properties. Neutrophils were cultured in either Veh (DMSO) or Ful (Fulvestrant) for 24 hours and RNA was harvested. Our results showed that there was no significant difference in mRNA expression of Elane (Figure 7a), Cd274 (Figure 7b), Tgfb1 (Figure 7c), and S100a9 (Figure 7d) (p > 0.05 for all). Some of the mRNAs showed a non-significant downward trend in the presence of Fulvestrant, an estradiol antagonist. Western blot showed a slight decrease in expression of ERα in the presence of Fulvestrant, however, these results lack a loading control (Figure 7e).
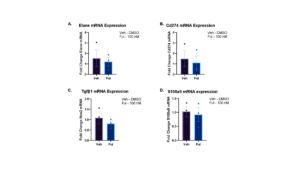
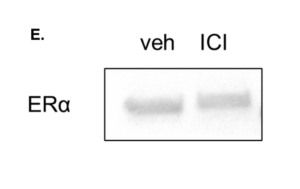 Figure 7. mRNA expression is not affected by Estradiol Antagonist Fulvestrant in Elane, Cd274, Tgfb1, and S100a9. Neutrophils in DF8 were cultured for 24 hours with either DMSO or Fulvestrant. RNA was collected and qPCR was run. For each experiment, 3 biological replicates were done and 2 technical replicates were run for qPCR. The data plotted is represented as ± SEM for Elane (a), Cd274 (b), Tgfb1 (c), and S100a9 (d). Unpaired t-test determined the significance between conditions with and without the presence of estrogen receptor α. A western blot was also run to see whether the expression of ERα decreased in the presence of Fulvestrant (e).
Figure 7. mRNA expression is not affected by Estradiol Antagonist Fulvestrant in Elane, Cd274, Tgfb1, and S100a9. Neutrophils in DF8 were cultured for 24 hours with either DMSO or Fulvestrant. RNA was collected and qPCR was run. For each experiment, 3 biological replicates were done and 2 technical replicates were run for qPCR. The data plotted is represented as ± SEM for Elane (a), Cd274 (b), Tgfb1 (c), and S100a9 (d). Unpaired t-test determined the significance between conditions with and without the presence of estrogen receptor α. A western blot was also run to see whether the expression of ERα decreased in the presence of Fulvestrant (e).
Discussion
Estrogen is a well-known stimulatory signal for some cancers. Prior studies have shown that estrogen is involved in the progression of multiple types of cancers including prostate, breast, and endometrial cancer in vivo and in vitro [26-30]. For example, cervical cancer patients with elevated estradiol levels were shown to have shorter progression-free survival rates and lower overall survival rates [26]. As mentioned earlier, LAM is an established estrogen-sensitive disease as demonstrated through multiple in vivo and in vitro studies. Estrogen has been shown to enhance lung colonization of TSC2-null ELT3 cells when mice were injected with tumor cells through the tail vein [5]. Furthermore, estrogen removal in a uterine-specific TSC2-null mouse model caused LAM-like tumors to shrink [6].
While in vivo studies involving TSC2-null cell lines have shown some transcriptional changes with estrogen, estrogen had minimal effects on cell proliferation, migration, and invasion, indicating that estrogen does not have as much of an effect in vitro as in the in vivo studies mentioned above [3, 6, 31]. To explain these observations, it is hypothesized that estrogen supports LAM disease progression by changing the function of immune cells in the disease setting. Our previous work showed that estrogen treatment in C57BL/6J mice increased both the presence of neutrophils and neutrophil elastase, a serine protease that promotes tumor growth and pro-inflammatory activities [32]. Furthermore, immunotyping of WT and uterine-specific TSC2-null mice revealed increased neutrophil accumulation in blood, bone marrow, lungs, spleen, and uteri, suggesting the LAM microenvironment is filled with neutrophils [3, 11]. Finally, our lab showed that estrogen alters the neutrophil phenotype and changes gene expression to promote tumor progression in LAM. [3]. These findings emphasize the importance of studying how estrogen signaling can influence the LAM microenvironment, and as a result, tumor growth and metastasis.
In this study, we proposed ERα signaling and tumor-derived factors promote a pro-tumorigenic, immunosuppressive neutrophil phenotype in the setting of LAM. We chose to measure specific mRNA markers because the proteins they encode have both immunosuppressive and pro-tumorigenic properties, and their expression in neutrophils would support our hypothesisUltimately, we found that Elane (Neutrophil Elastase), and Nos2 (Nitric Oxide Synthase 2) expression could depend on signaling through ERα. Since Elane and Nos2 are pro-inflammatory enzymes, this study shows that estradiol through ERα signaling could promote neutrophils to express inflammatory enzymes. Tumor-derived factors may have an effect as well, but this effect was not as prominent as ERα signaling for these two markers.
Cd274/PD-L1 (Programmed Cell Death Ligand 1) was found to be dependent on the tumor-derived factors in TCM. This dependence was further observed when we compared the values between tumor-conditioned media in the wild type and ERα-null mice. There was a clear difference in the levels of expression with and without estrogen receptors. Since PD-L1 is an immunosuppressive marker, immunosuppression could be better regulated by tumor-derived factors rather than ERα signaling.
Interestingly, the marker Cxcr2 indicates that estrogen signaling suppresses the full expression of these markers in both DF8 and TCM conditions. Estradiol may suppress neutrophil migration in response to chemokines as an activated neutrophil does not need to migrate since it is differentiated.
The markers Tgfb1, S100a9, and Irf8 were not affected much by estrogen signaling, but we saw that culturing the neutrophils in tumor-conditioned media increased the expression of these markers. Adding fulvestrant to neutrophil cultures did not significantly change mRNA expression for all of the probes ran (Elane, Cd274, Tgfb1, S100a9) which suggests that inhibiting ERα for a day in culture is not enough to affect mRNA expression as opposed to a mouse that has ERα knocked out. However, some results can be attributed to human error in running the qPCR protocol and pipetting errors. Neutrophil yields were not always consistent across each harvest and some experiments only had two replicates per condition (DF8 vs. TCM)or three replicates depending on the neutrophil number.
This study has demonstrated that ERα signaling between neutrophils and tumor cells potentially influences neutrophil phenotype and function by enhancing the expression of specific pro-tumorigenic genes. We have also shown that tumor-conditioned media, which contains tumor-derived factors, increases mRNA expression for multiple markers. These findings can serve as a direction for future treatments involving estrogen and estrogen signaling depletion to stop tumor progression or prevent crosstalk between neutrophils, estrogen, and LAM tumors. These findings could also be applied to other diseases involving estrogen or neutrophil-dependent tumors.
Future directions for this study include running bulk RNA sequencing on neutrophils to determine all the genes that may be upregulated in the presence of ERα signaling or tumor-conditioned media, as in this study we only chose a few due to time and cost constraints. Bulk RNA sequencing would elucidate the transcriptional landscape which would help us define the neutrophil phenotype stimulated by TCM and ERα signaling. Since an increase in mRNA doesn’t necessarily correlate with an increase in protein production, it would be beneficial to determine protein production. Quantifying protein products can be achieved through a combination of multiplex assays to capture a wider array of proteins. This can be further expanded upon through bulk RNA sequencing. The genes identified through qPCR can also be quantified with direct ELISAs. Another future step would be to determine what tumor-derived factors are in tumor-conditioned media to learn how they may enhance mRNA expression. Broad assays to investigate protein function can be done, as well as lipid sequestration to narrow down the possible origin of what factors are stimulating the mRNA expression in neutrophils. This would have to culminate in mass spectrometry to give us specific targets to look at. Finally, we should re-run the ERα western blot with β-Actin as a loading control to see if it was downregulated. Overall, we found that ERα signaling and tumor-derived factors affect the mRNA expression of neutrophils, promoting an immunosuppressive and pro-tumorigenic phenotype in the setting of LAM.
References
- Gibbons, E., B.M.N. Minor, and S.R. Hammes, Lymphangioleiomyomatosis: where endocrinology, immunology and tumor biology meet. Endocr Relat Cancer, 2023. 30(9).
- Johnson, S.R., Lymphangioleiomyomatosis. Eur Respir J, 2006. 27(5): p. 1056-65.
- Minor, B.M.N., et al., Estradiol Augments Tumor-Induced Neutrophil Production to Promote Tumor Cell Actions in Lymphangioleiomyomatosis Models. Endocrinology, 2023. 164(6).
- Harari, S., O. Torre, and J. Moss, Lymphangioleiomyomatosis: what do we know and what are we looking for? Eur Respir Rev, 2011. 20(119): p. 34-44.
- Yu, J.J., et al., Estrogen promotes the survival and pulmonary metastasis of tuberin-null cells. Proc Natl Acad Sci U S A, 2009. 106(8): p. 2635-40.
- Prizant, H., et al., Estrogen maintains myometrial tumors in a lymphangioleiomyomatosis model. Endocr Relat Cancer, 2016. 23(4): p. 265-80.
- Gu, X., et al., Integration of mTOR and estrogen-ERK2 signaling in lymphangioleiomyomatosis pathogenesis. Proc Natl Acad Sci U S A, 2013. 110(37): p. 14960-5.
- Adachi, K., et al., Intracrine steroid production and mammalian target of rapamycin pathways in pulmonary lymphangioleiomyomatosis. Hum Pathol, 2015. 46(11): p. 1685-93.
- Du, Y., et al., Lymphangioleiomyomatosis (LAM) Cell Atlas. Thorax, 2023. 78(1): p. 85-87.
- Guo, M., et al., Single-Cell Transcriptomic Analysis Identifies a Unique Pulmonary Lymphangioleiomyomatosis Cell. Am J Respir Crit Care Med, 2020. 202(10): p. 1373-1387.
- Taya, M., et al., Neutrophil elastase from myeloid cells promotes TSC2-null tumor growth. Endocr Relat Cancer, 2020. 27(4): p. 261-274.
- Lerman, I. and S.R. Hammes, Neutrophil elastase in the tumor microenvironment. Steroids, 2018. 133: p. 96-101.
- Galani, I.E. and E. Andreakos, Neutrophils in viral infections: Current concepts and caveats. J Leukoc Biol, 2015. 98(4): p. 557-64.
- Liew, P.X. and P. Kubes, The Neutrophil’s Role During Health and Disease. Physiol Rev, 2019. 99(2): p. 1223-1248.
- Hedrick, C.C. and I. Malanchi, Neutrophils in cancer: heterogeneous and multifaceted. Nat Rev Immunol, 2022. 22(3): p. 173-187.
- Mondal, S., et al., Matrix metalloproteinase-9 (MMP-9) and its inhibitors in cancer: A minireview. Eur J Med Chem, 2020. 194: p. 112260.
- Thomas, D.D. and D.A. Wink, NOS2 as an Emergent Player in Progression of Cancer. Antioxid Redox Signal, 2017. 26(17): p. 963-965.
- Cha, J.H., et al., Mechanisms Controlling PD-L1 Expression in Cancer. Mol Cell, 2019. 76(3): p. 359-370.
- Mollinedo, F., Neutrophil Degranulation, Plasticity, and Cancer Metastasis. Trends Immunol, 2019. 40(3): p. 228-242.
- Masugi, Y., et al., Tumour CD274 (PD-L1) expression and T cells in colorectal cancer. Gut, 2017. 66(8): p. 1463-1473.
- Korbecki, J., et al., CXCR2 Receptor: Regulation of Expression, Signal Transduction, and Involvement in Cancer. Int J Mol Sci, 2022. 23(4).
- Delobel, P., et al., CXCR2 intrinsically drives the maturation and function of neutrophils in mice. Front Immunol, 2022. 13: p. 1005551.
- Wang, J., H. Xiang, Y. Lu, and T. Wu, Role and clinical significance of TGF‑beta1 and TGF‑betaR1 in malignant tumors (Review). Int J Mol Med, 2021. 47(4).
- Markowitz, J. and W.E. Carson, 3rd, Review of S100A9 biology and its role in cancer. Biochim Biophys Acta, 2013. 1835(1): p. 100-9.
- Salem, S., D. Salem, and P. Gros, Role of IRF8 in immune cells functions, protection against infections, and susceptibility to inflammatory diseases. Hum Genet, 2020. 139(6-7): p. 707-721.
- Kozasa, K., et al., Estrogen stimulates female cancer progression by inducing myeloid-derived suppressive cells: investigations on pregnant and non-pregnant experimental models. Oncotarget, 2019. 10(20): p. 1887-1902.
- Dobbs, R.W., et al., Estrogens and prostate cancer. Prostate Cancer Prostatic Dis, 2019. 22(2): p. 185-194.
- Shang, Y., Hormones and cancer. Cell Res, 2007. 17(4): p. 277-9.
- Chung, S.H. and P.F. Lambert, Prevention and treatment of cervical cancer in mice using estrogen receptor antagonists. Proc Natl Acad Sci U S A, 2009. 106(46): p. 19467-72.
- Brake, T. and P.F. Lambert, Estrogen contributes to the onset, persistence, and malignant progression of cervical cancer in a human papillomavirus-transgenic mouse model. Proc Natl Acad Sci U S A, 2005. 102(7): p. 2490-5.
- Sun, Y., et al., Progesterone and estradiol synergistically promote the lung metastasis of tuberin-deficient cells in a preclinical model of lymphangioleiomyomatosis. Horm Cancer, 2014. 5(5): p. 284-98.
- Dai, R., et al., Neutrophils and neutrophil serine proteases are increased in the spleens of estrogen-treated C57BL/6 mice and several strains of spontaneous lupus-prone mice. PLoS One, 2017. 12(2): p. e0172105.
- den Boon, J.A., et al., Molecular transitions from papillomavirus infection to cervical precancer and cancer: Role of stromal estrogen receptor signaling. Proc Natl Acad Sci U S A, 2015. 112(25): p. E3255-64.
- Svoronos, N., et al., Tumor Cell-Independent Estrogen Signaling Drives Disease Progression through Mobilization of Myeloid-Derived Suppressor Cells. Cancer Discov, 2017. 7(1): p. 72-85.
About the Author
Katie is a Take5 student majoring in Biology and East Asian Studies. She is interested in the intersection of cancer biology and immunology. Katie finds it fascinating how environmental factors can influence the immune system and how its responses vary from person to person. She has been working in the Hammes Lab (URMC, Endocrinology and Metabolism) since junior year, and spent a summer in working Rowe Lab (URMC, Pediatric Infectious Disease) as well.
Cite this Article
O’Leary, K., Gibbons, E., Minor, B., Henson, T., Hammes, S. R. (2024). Estrogen Receptor α (ERα) Signaling and Tumor Cell-Derived Factors Mediate Pro-Tumor Neutrophil Activation in the Setting of Lymphangioleiomyomatosis (LAM). University of Rochester, Journal of Undergraduate Research, 23(1). https://doi.org/10.47761/WZIH8024
JUR | Creative Commons Attribution 4.0 BY International License